

近日,中国科学院分子细胞科学卓越创新中心(生物化学与细胞生物学研究所)徐国良研究组和季红斌研究组合作,以Loss of TET reprograms Wnt signaling through impaired demethylation to promote lung cancer development为题在PNAS上在线发表最新研究成果。通过对公共临床数据的深入分析并结合一系列临床前的自发成瘤小鼠模型,该研究揭示了DNA双加氧酶TET家族蛋白调控肺上皮细胞癌变过程中DNA甲基化的动态平衡,维持肺腺癌(lung adenocarcinoma, LUAD)发生发展中关键致癌信号通路Wnt signaling的拮抗基因的低甲基化及适当的表达,以确保Wnt signaling不被过度活化,进而抑制肺上皮细胞的癌前病变及肺腺癌的恶性进展。
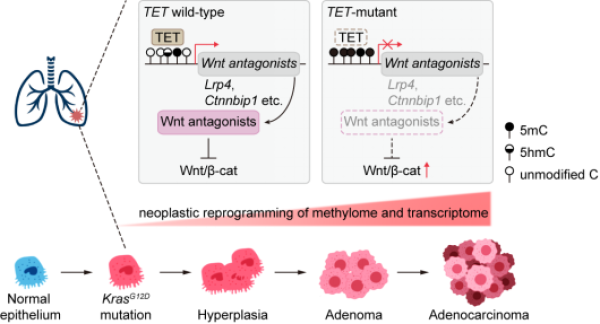
TET酶拮抗Kras驱动的肺腺癌发生发展的工作模型
徐国良团队长期致力于TET双加氧酶及其介导的DNA氧化去甲基化的分子机理及生物学功能研究(Xu et al., Nat Chem Biol, 2020)。TET酶能够迭代氧化5-甲基胞嘧啶(5mC),依次产生5-羟甲基胞嘧啶(5hmC)、5-醛基胞嘧啶(5fC)和5-羧基胞嘧啶(5caC),而5caC能被DNA糖苷酶TDG特异识别并切除,随后偶联碱基切除修复实现DNA主动去甲基化。基于这条DNA主动去甲基化通路,该研究团队也深入探讨了其在小鼠早期胚胎发育、成体神经发生与认知、体细胞重编程及原肠运动中的生物学功能。
众所周知,表观遗传的异常存在于各种类型的癌症中,并且表观遗传改变经常与遗传改变协同作用驱动致癌表型,其中就包括DNA甲基化的失衡。鉴于TET家族基因在血液肿瘤中相对较高的突变频率,绝大部分研究都关注TET酶在这类癌症中的作用。相比之下,由于突变频率较低,TET酶在实体肿瘤中究竟发挥怎样的作用?与血液系统肿瘤中的功能有何异同?TET基因的突变或TET酶的功能缺失是否会在实体肿瘤发生发展中赋予癌细胞选择性的生长优势?这些问题都还缺乏研究。
为了回答这些问题,研究人员分析发现TET基因在肺癌、肠癌及皮肤癌中的突变频率较高。鉴于肺癌在全球范围内的发病率和致死率居高不下,研究人员选择将最主要的肺癌亚型肺腺癌作为研究对象。进一步分析显示,TET基因在7.4%的肺腺癌中发生突变,与致癌驱动基因KRAS存在共突变关系,与EGFR突变则是互斥关系,且KRAS和TET共突变的病人生存期明显缩短。此外,TET基因突变会导致其自身的表达和氧化酶活受损。伴随着肿瘤发展,TET基因的表达水平也逐渐降低,且TET低表达的肺腺癌患者的生存期也相对较短。
研究人员构建了通过滴鼻诱导KrasG12D激活及Tet基因敲除的自发成瘤小鼠模型,来模拟上述临床特征。有趣的是,致瘤表型分析显示同时失活三个Tet基因剧烈加速了KrasG12D驱动的肺腺癌发生发展,而Tet基因单敲除或杂合敲除的结果表明Tet家族不同基因具有协同抑癌的作用,并且其抑癌效应主要依赖于氧化酶活。
机制研究发现,Tet敲除诱发了恶性癌变前体细胞中DNA甲基化和转录的重编程,尤其表现为Wnt signaling异常上调,同时呈现出Wnt signaling相关基因的高甲基化。在KrasG12D; Tet三敲除的肺腺癌细胞系中的回补实验显示,TET氧化酶活的恢复介导了Wnt相关拮抗基因(Lrp4, Ctnnbip1, Dact1, Tmem88)的启动子区域的氧化去甲基化,并重新激活了这些拮抗基因的表达,从而抑制了Wnt signaling过度活化。值得注意的是,这些小鼠模型中的发现也得到了临床数据的进一步支持,在TET突变的肺腺癌样本中WNT拮抗基因的高甲基化与其低表达相关。相应地,敲除Wnt signaling效应因子β-catenin,能够大幅度缓解由Tet缺失引发的肺癌表型,印证了Wnt signaling是受到TET酶氧化酶活调控的下游靶通路。
综上,该研究首次在实体肿瘤中建立了能够真实模拟临床的TET条件性敲除的自发肺腺癌小鼠模型,提供了Kras和Tet基因协同突变诱发恶性肺腺癌的遗传学证据,阐明了肺腺癌发生发展中表观遗传层面的重要调控机理,为开发针对TET突变的肺腺癌患者靶向抑制Wnt signaling的治疗方式奠定了理论基础。
该研究工作得到国家重点研发计划、国家自然科学基金、中科院战略性先导科技专项、中科院基础前沿科技专项、中科院国际合作项目等的经费支持。