

基因组中的DNA需要蛋白质的协助来完成编码、复制、重组和修复等过程。因此,绘制蛋白质与DNA相互作用的位置和方式,可以了解它们在生理和病理情况下如何发挥作用。
目前,已有研究团队开发出多种用于绘制单个靶蛋白与DNA全基因组相互作用的方法,包括DamID、染色质免疫沉淀测序(ChIP-seq)、CUT&RUN等。这些方法通过富集等方式选择性放大来自特定蛋白结合区域的DNA片段,并结合新一代测序技术(NGS)解析DNA序列,并将这些序列映射到参考基因组,使用测序覆盖率作为蛋白质- DNA相互作用频率的衡量标准。
虽然上述方法在研究DNA结合蛋白和染色质修饰方面非常有用,但也存在一些局限性。例如,常用的NGS技术通常为短读长,低于250个碱基对,这会影响那些长片段结合信息的正确解读。此外,人类基因组的重复区域对基因组组装和作图提出了重大挑战,因为很难非常准确地将短DNA测序片段比对到基因组中的独特位置。
为了解决上述挑战,加州大学伯克利分校研究团队开发了一种全新的蛋白质-DNA互作研究方法DiMeLo-seq(directed methylation with long-read sequencing, DiMeLo-seq)。DiMeLo-seq是一种以高分辨率绘制蛋白质- DNA互作的单分子长读长测序技术,并可同时检测内源性DNA修饰和序列变异,可提供多维度的全基因组信息,为基因组研究提供了前所未有的精度和广度。该研究发表在Nature Methods上,文章题为“DiMeLo-seq: a long-read, single-molecule method for mapping protein–DNA interactions genome wide”。

文章发表在Nature Methods
主要研究内容
DiMeLo-seq工作流程
据文章介绍,DiMeLo-seq结合了抗体导向的蛋白-DNA常规检测方法的元素,在特定的靶蛋白附近富集甲基化标记,然后使用长读长测序技术直接读取外源性甲基化标记。考虑到人体内DNA N6 -甲基脱氧腺苷(mA)的低丰度,研究人员将抗体结合蛋白A与非特异性脱氧腺苷甲基转移酶Hia5融合(pA-Hia5),在靶向染色质相关蛋白的DNA近端催化mA的形成。
DiMeLo-seq方法在检测目标蛋白在每个长单DNA分子上的多个结合事件方面具有独特优势 ,且这是短读长测序不可能实现的。此外,DiMeLo-seq方法还允许同时检测外源性抗体导向的腺嘌呤甲基化和内源性的单分子CpG甲基化。更重要的是,长读长测序可以在高度重复的基因区域内绘制蛋白质-DNA相互作用的图谱。
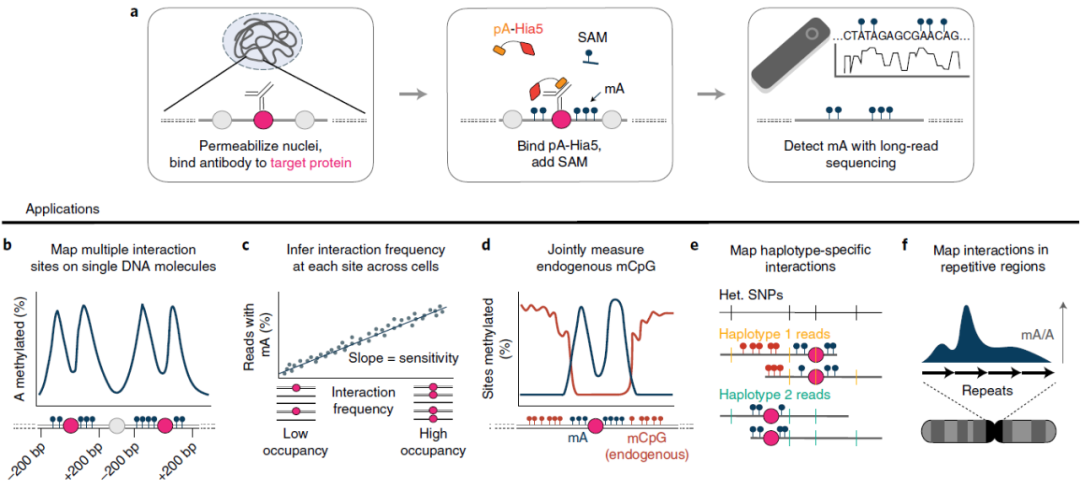
图1. DiMeLo-seq工作流程图,来源:Nature Methods
单分子CTCF结合和CpG甲基化的联合分析
DiMeLo-seq具有同时检测内源性CpG甲基化、核小体占用和蛋白结合来表征局部染色质环境下蛋白质-DNA互作的能力。研究团队以CTCF为靶点(CTCF是一种主要定位于核小体周围的蛋白,其结合可被CpG甲基化抑制),以标准化的ChIP -seq数据为参考,进一步验证了DiMeLo-seq与ChIP-seq数据的一致性,并可视化蛋白质在单个分子上的结合,发现DiMeLo-seq信号随ChIP-seq信号强度的变化而变化。
研究团队探讨了CTCF结合与内源性CpG甲基化的关系。分析结果表明,CTCF结合位点在motif周围的mCpG中大幅下降,在较弱的ChIP-seq峰中下降较少,这一CpG甲基化和CTCF靶向甲基化的反比关系反映mCpG可抑制CTCF的结合。
此外,研究人员还研究了DiMeLo-seq在检测单个分子上蛋白结合相邻位点的能力。通过在两个单分子结合位点上分析CTCF的结合特征发现,DiMeLo-seq能够检测到相邻CTCF基序可与两个位点或两个位点中的一个结合,证明DiMeLo-seq在分析长单分子上的协调结合模式方面的潜力。更重要的是,在25X的覆盖率下,DiMeLo-seq能够检测到同一单个分子上两个位点的结合模式,并将这两个位点中缺失的结合模式归因于结合基序内的突变。上述结果表明,DiMeLo-seq可以检测单倍型特异性遗传或表观遗传变异对蛋白质结合的影响。
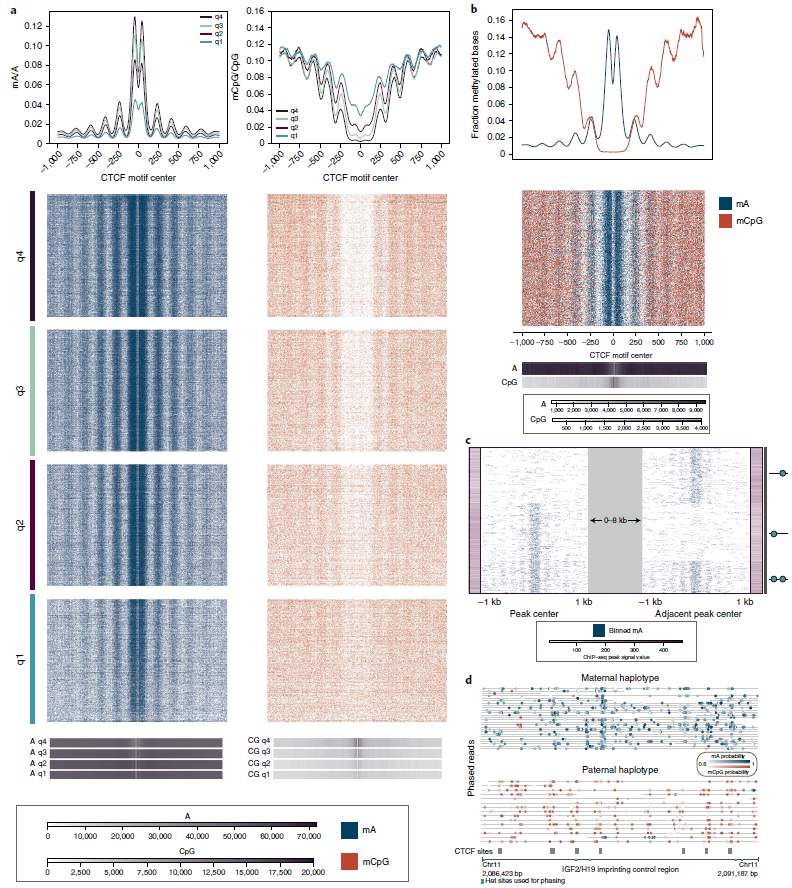
图2.单分子CTCF结合和CpG甲基化的图谱,来源:Nature Methods
利用DiMeLo-seq定位异染色质组蛋白修饰
为了验证DiMeLo-seq测量基因组异染色质重复区域内蛋白质结合的能力,研究团队选择HG002细胞中的H3K9me3为靶点,因为这个男性来源的淋巴母细胞细胞系的X染色体着丝粒已经完全组装好,且含有大量H3K9me3。为了验证靶向甲基化的特异性,研究团队计算了CUT&RUN H3K9me3峰内甲基化腺嘌呤的比例,并与广义峰外甲基化腺嘌呤的比例进行了比较。分析发现,在CUT&RUN峰中mA/A的富集程度比背景高3.6倍,表明在预期的基因组中H3K9me3区域内DiMeLo-seq可检测到甲基化富集。
为了验证H3K9me3指向的mA信号在着丝粒中的富集,研究团队同样计算了mA/A的富集倍数,也发现在着丝粒中富集了约1.9倍。
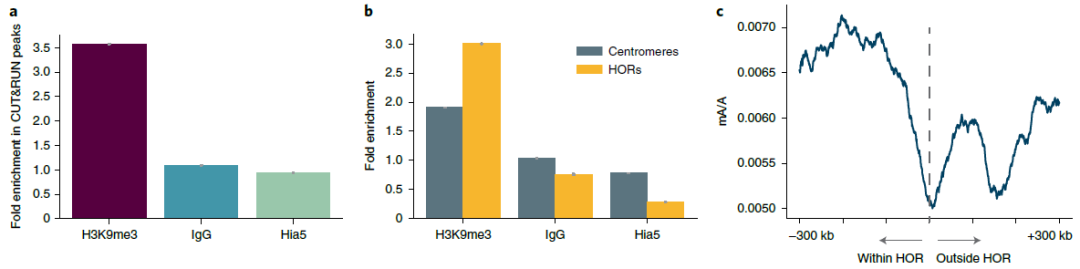
图3.着丝粒区域中H3K9me3的表征,来源:Nature Methods
结 语
该研究团队开发、优化并验证了DiMeLo-seq,并证实是一种全基因组范围内绘制蛋白质-DNA互作的长读长测序方法。DiMeLo-seq可以在数百个碱基对内绘制一个蛋白质的结合位点,并可在单个DNA分子上检测多个位点。这种长读长测序技术提高了基因组高度重复区域的可研究性,为未来研究相关调控和功能打开了思路。
同时,由于DiMeLo-seq不涉及扩增,因此可以用来更好地估计基因组中每个位点的蛋白质-DNA互作的绝对频率。DiMeLo-seq还提供了内源性CpG甲基化和蛋白质-DNA互作的信息,可以分阶段揭示单倍型特异性结合和甲基化模式。总之,DiMeLo-seq提供了一种强大的方法来描述全基因组范围内的蛋白质-DNA互作。