

导 读
癌症是一个逐渐演化的病理过程,持续的基因突变导致了肿瘤细胞群内的遗传多样性。虽然癌症中的每个细胞在本质上都是不同的,但通常也存在基因组几乎相同的细胞群,即所谓的克隆群体。克隆之间的进化关系可以用系统发育树或克隆树来表示。在过去十年中,从序列数据推断克隆群体结构、基因型和发育树一直是个活跃的研究领域。
早期的克隆分析方法基于混合样本的序列数据,通过计算反卷积来解决数据的混合性质。近年来,人们发现将单细胞DNA测序(scDNA-seq)数据与Bulk混合测序数据集成进行联合分析能够显著提高准确性、解析克隆群体结构,但其无法识别基因组异质性导致的功能差异。
近期,科研人员开发了“两步法”,即识别克隆群体结构;将单细胞RNA测序(scRNA-seq)数据与克隆基因型对齐。但两步法不能充分利用现有数据,因此,目前亟需一种综合方法来同时从大量混合DNA-seq和scRNA-seq数据中识别克隆群体结构和相关克隆基因型,以揭示克隆基因表达谱中的肿瘤异质性。
近日,瑞典斯德哥尔摩KTH皇家理工学院、加拿大不列颠哥伦比亚大学的科研人员在Nature Communications发表了题为“Reconstructing clonal tree for phylo-phenotypic characterization of cancer using single-cell transcriptomics”的文章。研究团队介绍了一种名为PhylEx(Phylo Expression)的贝叶斯统计方法,其集成了DNA-seq和scRNA-seq数据,可利用在单细胞内观察到的单核苷酸变异(SNV)信息来识别克隆、改善克隆树重建,并将RNA表达谱信息高精度映射到克隆上。PhylEx解锁了系统表型分析的潜力,能够在与克隆基因型相关的单个克隆基因和通路水平上发现、描述肿瘤的进展及复发机制。

文章发表在Nature Communications
主要研究内容
将scRNA与DNA数据集成可以改进克隆树的重建
PhylEx是一种贝叶斯统计方法,在重建克隆树同时,可为以Bulk DNA-seq和scRNA-seq数据为特征的肿瘤克隆分配单细胞和基因型。PhylEx能够从克隆树的后验分布以及最大后验(MAP)树上生成样本,输出克隆基因型及细胞-克隆分配。
在无拷贝数畸变的进化模型下,研究团队在樱桃形树上模拟了100个SNV和20个单细胞的Bulk和scRNA-seq数据。使用PhylEx对数据进行分析,并将其与基于Bulk的克隆树重建方法PhyloWGS进行了比较。总体而言,两种方法均能正确地推断细胞的克隆演化进程,但PhyloWGS推断出的是线形树,相比之下,PhylEx通过利用单细胞数据中突变的共现,正确地推断出樱桃形树,并对SNV和细胞进行共聚类。
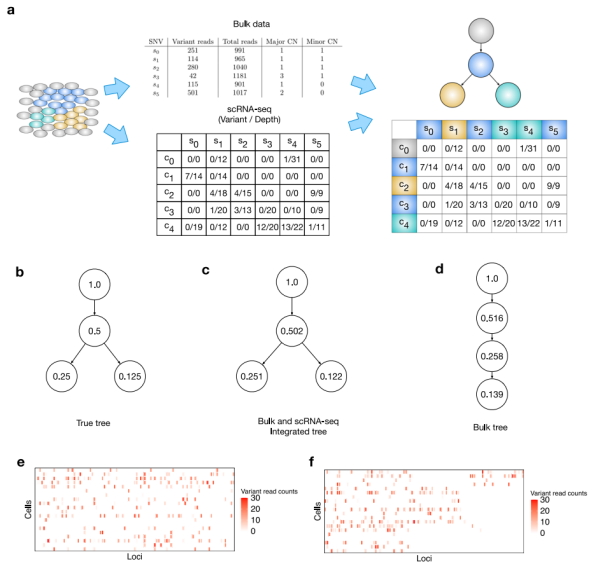
图1. PhylEx的工作原理。来源:Nature Communications
PhylEx通过集成数据重建高保真克隆树
多区域测序是提高克隆树重建准确性的标准方法。研究团队评估了PhylEx在由单区域Bulk DNA-seq和scRNA-seq数据组成的模拟数据上的性能,并将其与提供多区域DNA数据的Bulk方法之间的性能进行了比较。
在不考虑拷贝数演化和给定多区域数据的情况下,Bulk方法实现了较高的精度,但当提供单细胞数据时,PhylEx表现更好。在拷贝数进化模型下模拟数据时,即使提供多区域数据,Bulk方法也很难重建克隆树。相反,PhylEx利用scRNA-seq和单区域Bulk DNA-seq数据,能够重建准确性较高的克隆树,这增强了克隆树重建方法在各种仅限于单区域测序的研究、临床环境中的适用性。
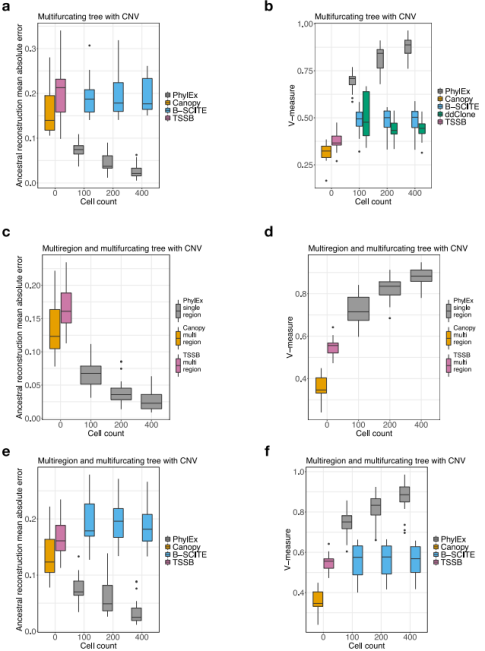
图2. PhylEx的性能评估。
PhylEx重建浆液性卵巢癌克隆谱系
为评估PhylEx在真实数据上的性能,研究团队分析了一组相关的高级别浆液性卵巢癌细胞系。这些细胞系来自同一病人,一个来自原发肿瘤(OV2295),两个来自复发标本 (OV2295R2和TOV2295R)。研究团队使用直接文库制备(DLP)scDNA-seq技术对上述细胞系进行了检测,并由计算肿瘤学领域的知名专家使用多种基因组特征进行分析以重建克隆树,将其称之为“DLP克隆树”。研究团队对DLP克隆树与由PhylEx重建的克隆树进行了比较分析,以评估PhylEx在实际测序数据上的性能。
结果显示,DLP克隆树与PhylEx克隆树具有很强的一致性,具有几乎相同的拓扑结构;PhylEx正确分配了24个祖先突变中的23个。在三个聚类指标和祖先重建指标上,研究团队比较了PhylEx与Canopy、TSSB、ddClone和B-SCITE等克隆重建方法的推断结果。结果显示,PhylEx的性能明显优于其他所有方法。
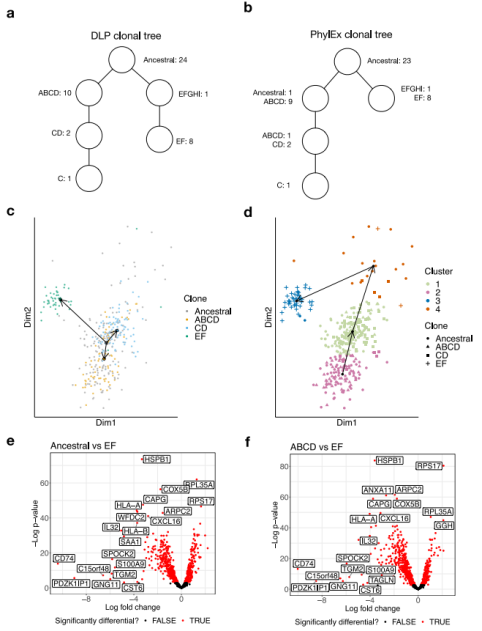
图3. 卵巢癌克隆谱系的分析。
解析HER2+乳腺癌的表型进化
研究团队在未经治疗的HER2+乳腺癌肿瘤的五个空间不同区域生成了Smart-Seq3 scRNA-seq和Bulk全外显子组DNA测序数据,将PhylEx应用于预处理后可用的369个细胞和418个SNV上。结果显示,PhylEx构建的MAP树是线性扩展(即一条路径);在PhylEx树上分配的细胞形成了清晰的分区,错误区域较小。
研究团队还检索了NanoString人类泛癌相关通路的770个基因用于下游分析,以识别每个克隆的驱动突变。在祖克隆(克隆1)中,研究团队发现了CDC6的突变,这意味着细胞复制机制的改变;在克隆2中发现了TP53和MAP3K8的突变,这表明癌症从克隆2开始扩散;在克隆3中,涉及PI3K和MAPK通路的基因(PIK3R3、CACNA2D2)和MDC1(DNA修复)发生突变;克隆4以RAS通路的突变为特征。由此可见,克隆树为分析和检查癌症突变提供了一个重要的背景。
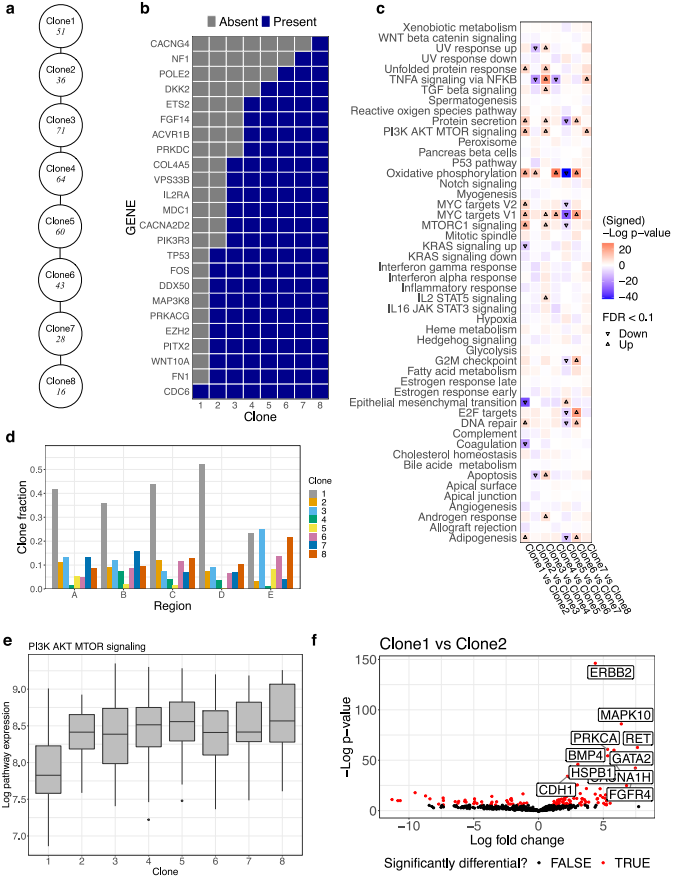
图4. 多区域HER2+乳腺癌分析。
最后,研究团队对MSigDB Hallmark基因集进行了基因集富集分析,结果显示,克隆2中PI3K - AKT MTOR信号通路(乳腺癌中常用的活化治疗靶点)的表达明显高于克隆1;克隆1的所有克隆中PI3K - AKT - MTOR信号通路均上调。此外,与克隆1相比,克隆2中ERBB2存在显著过表达。
上述结果表明,PhylEx分析识别了驱动突变,阐明了克隆的空间分布,并促进了scRNA表达数据的下游分析,揭示了克隆的功能特征。
结 语
综上所述,该研究提出了PhylEx方法,其可整合基因组和单细胞转录组数据来重建克隆树,这为通过系统表型分析来描述单个克隆的功能状态提供了新的思路。与现有的克隆重建方法相比,PhylEx性能更优。此外,基于真实数据的分析,PhylEx揭示了进化过程和克隆表型之间的相互作用。随着DNA-seq的流行及scRNA-seq研究的不断扩大,PhylEx将为研究癌症进化功能影响的癌症研究人员带来更多便利。