

去年,中国食品药品检定研究院王军志院士联合了同济大学附属东方医院等单位,在知名期刊《Signal Transduction and Targeted Therapy》上发表了一篇题为《Extracellular vesicle-based drug overview: research landscape, quality control and nonclinical evaluation strategies》的综述。1在这篇综述中,王院士站在一个全景视角的高度,为我们详细描绘了细胞外囊泡的成药路径,起到了全行业科普与路线图指引的作用。就在昨天,中检院再度出击,在细胞外囊泡领域的顶刊《Journal of Extracellular Vesicles》上面刊登了一篇“致编辑信(LtE)”。2中检院细胞室的纳涛副研究员、张可华副研究员为这篇文章的共同第一作者,孟淑芳主任和国家药监局药品评价中心张辉主任为通讯作者,来自北京恩泽康泰的赵立波博士也参与了部分撰写工作。这篇文章虽然不算正式的监管指南,但是作为一个面向中国干细胞来源EV治疗产品的质量评价框架建议,同样具有“里程碑”式的意义。相比王院士的上一篇综述,这篇JEV高度聚焦于“干细胞衍生”的天然EV,死磕“药学与质量控制(CMC),不仅展现了极强的审评严谨性与风险意识,也向全球展示了中国在先进治疗产品(ATMP)监管科学领域的专业深度,有助于中国参与甚至主导未来国际标准(如ICH相关指南)的制定。下面小编站在一个细胞外囊泡从业者的角度,为大家进行解读一下这篇文章。

一、这篇文章为什么重要?
首先,细胞外囊泡(EV)领域长久以来存在一个很微妙的现象,就是学术界和产业界在“自说自话”。例如,国际细胞外囊泡学会(ISEV)发布MISEV指南的初衷是为了提高EV领域科研工作的“标准化”和“规范化”。一方面,这些研究并非仅限于EV治疗产品的开发,而是包括诊断、生物学机制研究在内的所有领域。另一方面,即便是围绕治疗方面的研究与应用,MISEV的关注点也更多地是侧重在诸如“你研究的是不是 EV?数据是否可被重复”这一类问题。而产业界和监管机构关心的则是“这个EV产品是否可定义、可生产、可放行、可控、可证明安全有效”
Ameya P. Chaudhari及其同事在《Nature Reviews Bioengineering》上发表的题为《The status of extracellular vesicles as drug carriers and therapeutics》的综述中就曾提到,目前EV治疗产品已发表的药效学研究存在较为严重的问题。例如,超过87%的研究结果没有进行剂量效应的研究,仅4%给出了IC50的数值。显然,大量已发表的药效学研究几乎不符合药物开发的标准,这无疑就是EV治疗产品基础研究和产业化应用之间严重割裂的现实写照。而这篇综述,系统性地梳理并明确了科研标准(MISEV)与药学标准(ICH规范、药典)的区别,将EV从一个“生物学概念”拉入了“药学概念”。将这两个层次区分清楚,对行业未来的良性发展非常重要。同时,务实的检测策略对于企业的商业化生产也具有很强的现实指导意义。
二、“EV药品质量评价”框架呼之欲出,又恰逢其时
去年,间充质干细胞(MSCs)治疗产品在中美两国先后获批,让从事干细胞药物研发的国内外企业看到了希望。但是截止目前,国内外都尚未出台专门针对EV药物的CMC(药学研究与质量控制)指导原则,大量处于临床前研发阶段的生物技术公司面临着“不知道向监管机构交什么数据”的困境。在这样的大背景下,这篇文章为国内EV类药物的临床试验(IND)申报提供了一个可供参考的“风向标”。
一方面,文章清晰地界定了七大维度的质量评价体系(从细胞库到纯度、理化特性、安全性等),相当于给企业提供了一份详尽的“考纲”。企业完全可以参照文中的表3和表4,对照检查自家产品的开发路径是否符合要求。
另一方面,中检院专家通过在国际顶级期刊发表文章的形式,向国内外学术界和产业界释放了中国监管机构对EV药物的审评底线和倾向。这不仅能够大幅降低企业的试错成本,无疑也会加速国内EV类药物从实验室走向IND的进程。
三、敲响了“无细胞疗法绝对安全”的警钟
在过往发表的很多研究中,干细胞衍生EV常被宣传为比活体干细胞更安全的“无细胞(Cell-free)”替代品,认为其没有致瘤风险或免疫排斥。但这篇文章展现了极强的审评严谨性与风险意识。作者深刻剖析了EV可能存在的安全盲区,例如携带致癌miRNA(如miR-21)引发的致瘤性,表面富含负电荷脂质可能引发的促凝血(血栓)风险和溶血风险,以及携带非自体抗原引发的免疫原性。同时,强调了要覆盖生产全程,并从源头控制开始的质量控制策略的必要性。打破了“只要末端提纯做得好就行”的侥幸心理,这促使研究人员和企业在开发早期就必须将安全性评估(毒理、致敏、凝血)纳入考量。
四、国际战略意义
数据显示,中国在EV领域的临床试验(含IIT)数量位居全球第二(占绝大比例的是MSC-EV研究),但是到目前为止国内尚无IND获批。而中国监管机构的专家在国际权威期刊(JEV)提出这套质量评价框架,不仅向国内生物技术公司表达了开放的态度,也向全球展示了中国在先进治疗产品(ATMP)监管科学领域的专业深度,有助于中国参与甚至主导未来国际标准(如ICH相关指南)的制定。此外,文章不仅分析了中国的情况,还对比了美FDA、欧EMA的监管现状,提出的框架具有很强的普适性。它不仅服务于中国,也为全球面临同样监管困境的卫生当局提供了极具参考价值的“中国方案”。可以说,这篇文章也是中国在跻身国际先进治疗药品开发和监管强国之路上擂响的又一声战鼓。
五、原文详细解读
接下来,让我们紧随原文内容,看看干细胞外泌体治疗产品如何从“能做出来”真正走向“可评价、可放行、可临床转化”!
1. 研究背景
细胞外囊泡(EV)是由脂质双层膜包裹、由细胞释放到胞外空间的纳米至微米级囊泡,不能自我复制,但可以携带蛋白质、脂质、核酸、代谢物和糖类等多种“货物”。这些货物往往保留了来源细胞的生物学特征,并通过细胞间通讯调节靶细胞功能,因此在组织修复、免疫调节、药物递送和疾病治疗中展现出巨大的应用潜力。
目前,干细胞来源EV,尤其是间充质干细胞(MSCs)来源EV,是治疗性EV产品开发中最受关注的方向之一。文章统计www.clinicaltrials.gov数据显示,截至2025年9月,中国已成为EV临床研究注册数量仅次于美国的国家;在161项中国机构注册的EV相关研究中,42项聚焦治疗性EV。2016年至2022年仅注册11项治疗性EV研究,而自2023年以来近三年数量已增长至此前的约3倍。
从来源看,已注册治疗性EV研究中,超过95%涉及天然干细胞来源EV,其中MSCs占比约60%;在MSCs来源EV中,脂肪来源约占12%,脐带来源约占44%,另有44%因注册信息不完整而无法进一步判断来源。地域上,治疗性EV研究主要集中在上海、广东和北京。
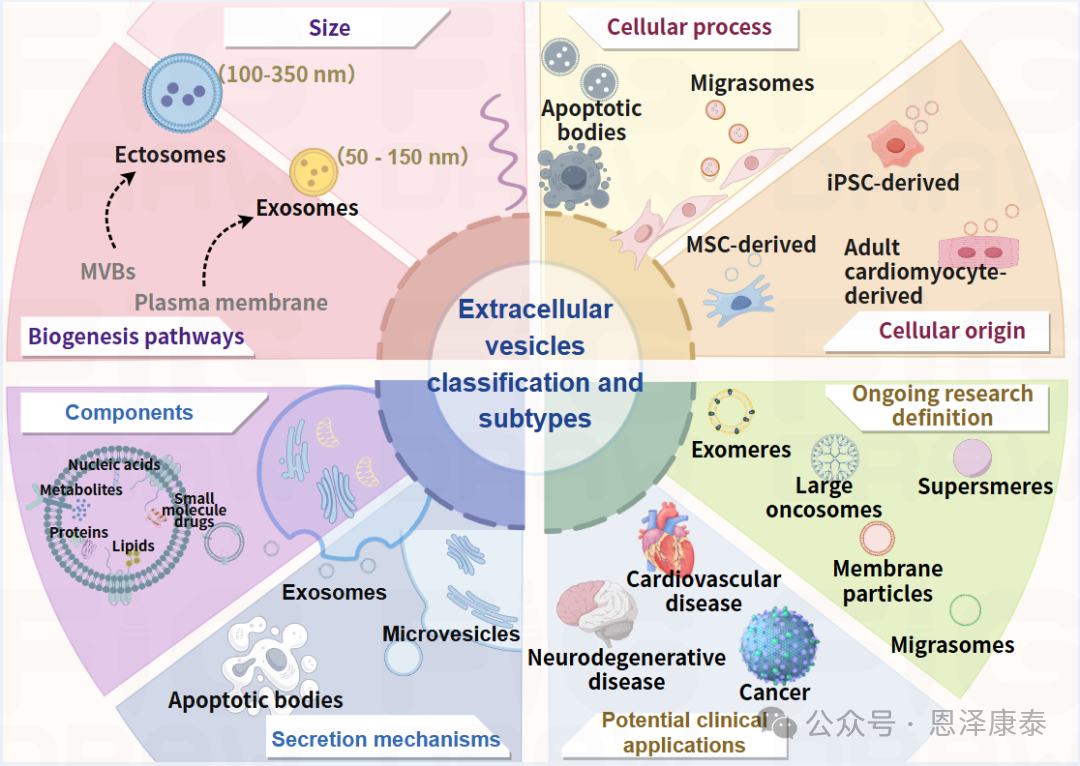
Fig. 1. 文章梳理了EV可根据大小与生物发生途径、细胞过程、细胞来源及胞内货物等不同维度进行分类。
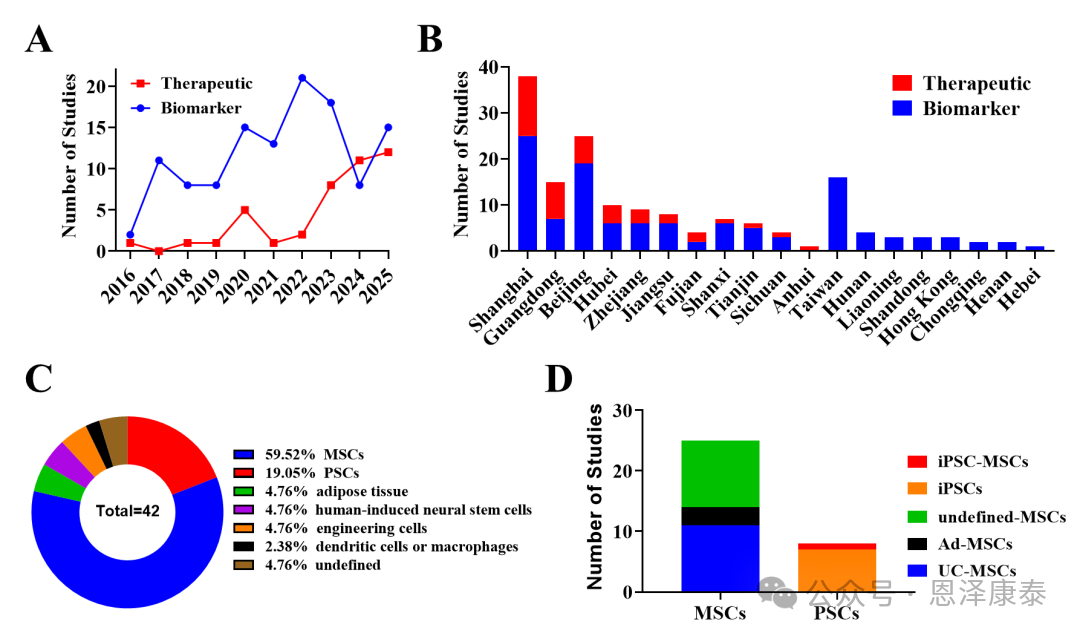
Fig. 2. 中国机构在ClinicalTrials.gov注册的EV临床研究概况,显示治疗性EV研究近年来增长明显,且干细胞来源EV占据主导。
与此同时,EV作为新型治疗产品的监管归属和质量评价体系仍在快速演进。2025年6月10日,国家药监局药审中心(CDE)发布《先进治疗药品范围、分类及释义(征求意见稿)》,明确EV属于先进治疗药品(ATMPs)的监管范围。此外,国家食品药品检定研究院也曾明确建议EV不宜按医疗器械进行管理。换句话说,EV治疗产品的核心问题,已经从“有没有外泌体”进入到“如何按药品逻辑证明其安全、有效、质量可控”。
2. 文章主要内容
本文章围绕干细胞来源EV治疗产品的全生命周期质量控制展开,将质量评价前移至细胞库建立阶段,并进一步延伸到EV分离纯化、理化表征、强度和效价、纯度、生物学功能、安全性、制剂稳定性、放行检测方法以及批间一致性等多个层面。
从框架上看,文章不是简单罗列检测项目,而是试图回答一个更关键的问题:哪些质量属性真正与EV产品的安全性、有效性和一致性相关,哪些检测方法适合研发阶段,哪些方法又能真正进入放行检测和产业化质量体系。
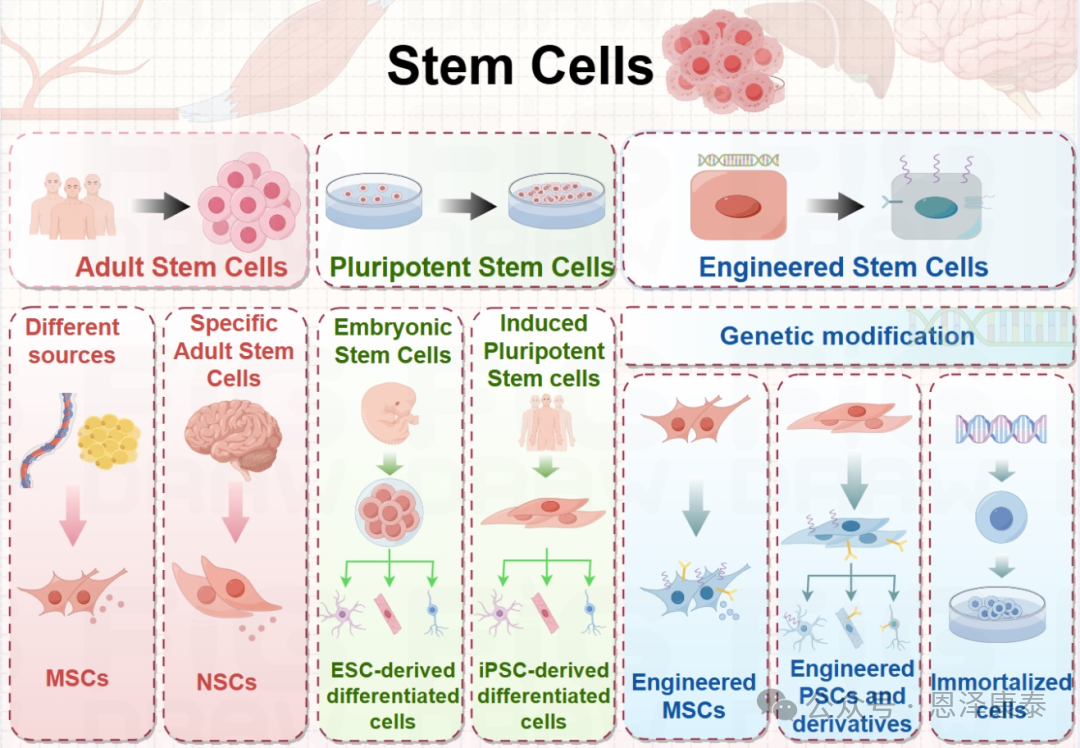
Fig. 3. 本文聚焦治疗性EV生产相关干细胞类型,包括MSCs、PSCs、PSC分化细胞以及基因修饰干细胞等。
关键内容1.细胞库不是配角,而是EV产品质量的源头
文章首先提出,干细胞应被视为EV生产的核心原材料。因为EV的成分和生物学功能会直接反映来源细胞的状态,细胞来源、培养体系、传代次数和遗传稳定性,都可能影响EV的质量和产量。
对于用于EV生产的MSCs,文章建议在建立细胞库前,就应评估其EV分泌水平、不同传代过程中的EV生产稳定性,以及是否适合用于EV产品开发。也就是说,适合细胞治疗的细胞来源,不一定天然适合EV治疗产品生产;EV产品需要自己的源头质量逻辑。
在细胞库质量控制方面,文章列出了身份鉴别、无菌、支原体、外源和内源病毒、细胞活性、促瘤性/致瘤性、基因组稳定性、残留物、基因编辑相关检测等多个项目。对于基因修饰细胞,还需要进一步关注插入位点、拷贝数、目的基因表达、编辑效率、脱靶效应以及这些改变是否会影响EV的组成和功能。
这部分内容的价值在于,它把EV治疗产品的第一个质控点提前到了细胞库阶段。只有来源细胞稳定、可追溯、风险可控,后续EV产品才有可能实现真正意义上的批间一致。
关键内容2. EV身份鉴定:不能只靠CD9、CD63、CD81“打卡”
目前很多EV研究仍主要参考MISEV2023推荐的通用标志物,如CD9、CD63、CD81以及部分胞质蛋白。这些指标对证明样本具有EV特征非常重要,但对于治疗产品而言,仅仅证明“这是EV”还不够。
文章特别指出,通用EV标志物往往跨细胞类型共享,无法充分说明供者来源、组织来源、细胞类型或基因修饰状态。而这些信息恰恰与EV产品的疗效和安全性密切相关。因此,治疗性干细胞来源EV应建立“通用标志物+来源特异性标志物”的双层身份评价体系。
例如,MSC来源EV可进一步考虑CD73、CD105、CD44等阳性特异标志物,以及CD11b、CD14、CD19、CD45、CD79等阴性标志物;PSC来源EV则已有研究提示PODXL、SSEA4等可作为特异性标志物。对于不同产品,还应结合工艺、来源细胞和适应症,逐步建立企业内部的标志物阳性率标准和放行阈值。
此外,文章还讨论了供者混池问题。混合多个供者细胞或EV有时会降低数据层面的波动,但也可能掩盖真实的供者差异,并引入免疫原性和可追溯性风险。因此,对于治疗产品,更应强调来自清晰、可追溯、充分表征的细胞来源,并通过STR等方法确认EV的细胞供体来源。
关键内容3. 理化性质:不只是看见“杯状结构”
在EV表征中,透射电镜(TEM)、扫描电镜(SEM)和冷冻电镜(cryo-EM)常用于观察EV形态。但文章提醒,传统电镜样品制备中的固定、脱水和真空条件,可能让EV呈现典型“杯状”形态,这并不一定完全反映其天然状态。因此,形态学评价的重点,不应只是看他形态像不像,而应关注脂质双层膜是否存在、膜结构是否完整。
粒径分布是另一个关键质量属性。治疗性EV常见粒径约为30-150 nm,但检测结果会受到分离方法、仪器平台、样本浓度、取样体积和EV亚群比例等多种因素影响。文章建议采用经验证的NTA、TRPS或nano-flow cytometry等方法,并结合纳米标准品和正交检测策略,提高结果的可靠性和可比性。
更重要的是,不同粒径亚群可能对应不同的生物功能。例如,某些小EV亚群在血管生成、抗纤维化或组织修复中可能更具活性,而大粒径亚群未必具有相同效果。因此,当粒径分布与特定生物功能相关时,特定粒径范围本身就应被纳入质量属性,而不是只报告一个“总颗粒数”。
文章还将Zeta电位纳入理化质量评价。Zeta电位与EV分散体系稳定性、聚集风险、表面相互作用以及潜在生物功能相关,可作为反映EV胶体稳定性的重要指标。
关键内容4. 强度、纯度与生物功能:从“多少颗粒”走向“多少有效颗粒”
对于EV治疗产品来说,剂量和强度的评价不能只看总颗粒数,也不能简单用总蛋白、总RNA或总脂质含量替代。文章提出,EV产品的“强度”可理解为每一定数量EV颗粒中所含特定活性成分的量,这些活性成分需要与产品预期生物功能或适应症建立明确关联。
例如,当某个miRNA、蛋白质或脂质组分被严谨验证与疗效相关时,其定量水平才有可能成为强度或效价相关指标。相反,如果仅报告“一共有多少蛋白”或“一共有多少RNA”,而没有功能学验证,就很难说明这些成分真正代表治疗活性。
纯度方面,文章提出应从多个角度综合判断:EV膜完整性、目标粒径范围内颗粒比例、携带活性成分颗粒比例、非目标细胞来源EV比例、蛋白和核酸污染、培养基或工艺残留等。特别是膜完整性评价非常关键,因为膜破裂可能导致内容物泄漏、功能下降,也可能暴露免疫活性成分,引发额外安全风险。
在生物学功能评价中,文章区分了“特异功能”和“一般功能”。特异功能应与目标适应症直接相关,例如免疫调节、促血管生成、组织修复、抗炎或抗凋亡等;一般功能则可作为辅助功能共同支撑产品整体作用。早期研发阶段应尽可能建立多维度功能数据库,随着机制逐步清晰,再筛选出与疗效最相关、可量化、可验证的质量控制指标。
关键内容5. 安全性评价:外泌体“不复制”,不代表没有风险
EV本身不能复制,很多人因此认为其天然更安全。但文章非常明确地指出,作为治疗产品,EV仍需要系统评估安全风险,主要包括EV内源成分风险、细胞培养与制备过程风险,以及储存、运输和使用过程中的风险。
成瘤性/致瘤性是首先需要关注的问题。文章以miR-21为例说明,某些EV携带的miRNA在组织修复中可能具有积极作用,但在另一种生物学背景下也可能与肿瘤发生、转移、血管生成或耐药相关。因此,对于兼具治疗效应和潜在风险的活性成分,不能只看“有没有”,还要建立安全阈值和有效剂量之间的边界。
凝血和溶血风险同样不能忽视。EV表面的负电荷磷脂,如磷脂酰丝氨酸(PS),可能为凝血反应提供催化表面,进而影响凝血级联反应;在特定疾病或易感人群中,这类风险可能进一步放大。
免疫原性方面,EV可能携带MHC分子、细胞因子、趋化因子、热休克蛋白、RNA等免疫活性分子,引发免疫识别、免疫激活、免疫清除甚至自身免疫反应。对于异体来源、PSC来源、基因修饰来源或永生化细胞来源EV,这部分评价尤其重要。
此外,文章还强调了残留风险,包括细胞培养、分离纯化和制剂过程中可能引入的溶剂、试剂、动物或人源添加物、外源蛋白、外源核酸、基因编辑材料等。这些成分如果去除不充分,可能带来细胞毒性、免疫反应或功能干扰。
关键内容6. 制剂与稳定性:EV产品不能只停留在“新鲜样本”阶段
治疗产品最终需要被储存、运输、给药和长期管理,因此制剂质量也是文章重点讨论的内容。目前EV制剂主要包括冻干制剂和液体制剂两类。EV在不合适的温度、pH、离子强度或反复冻融条件下,可能发生聚集、沉淀、膜破裂、蛋白降解、RNA降解或功能丢失。
为了提高稳定性,文章提到可考虑使用蔗糖、人血清白蛋白联合海藻糖、PLGA-PEG等保护剂或辅料,但这些辅料本身也必须进行兼容性和安全性评价。因为辅料可能影响粒径和颗粒数检测、膜完整性、表面蛋白状态甚至EV与靶细胞的相互作用。
对于冻干EV制剂,还需要确认冻干过程是否破坏EV结构和生物活性,复溶后是否能恢复原有功能,并观察是否存在可见沉淀、聚集或不溶性颗粒。对于需要缓释或控释的EV制剂,也应通过体外释放实验评价释放曲线和活性成分稳定性。
文章还提出,包装材料、容器密封完整性、运输条件、长期储存中的降解产物、批间一致性等,都应纳入整体质量控制体系。这也意味着EV产品开发不能只关注“分离出一管高纯度EV”,更要考虑它能否稳定地成为一种可使用、可运输、可放行的制剂。
关键内容7. 与MISEV2023的差异:从科研标准走向药品开发逻辑
MISEV指南是EV基础研究领域最重要的标准之一,核心价值在于规范术语、实验设计和EV表征,提高研究结果的严谨性与可重复性。本文并不是替代MISEV,而是在MISEV基础上,进一步面向治疗产品开发和监管评价,提出更接近药学研究逻辑的质量控制策略。
文章总结了两者之间的几个关键差异:
第一,目的不同。MISEV主要回答“研究对象是不是EV,以及作用是否由EV引起”;而治疗产品质量评价还要回答“是否安全、有效、可控、可放行”。
第二,身份鉴定更具体。治疗性EV产品不仅需要EV通用标志物,还需要来源细胞、组织来源、供者来源、基因修饰状态等更细分的信息。
第三,评价维度更完整。除基础表征外,还要加入促瘤性、免疫原性、凝血/溶血、残留物、制剂稳定性等与临床使用密切相关的项目。
第四,评价贯穿全生产过程。细胞库、培养上清、中间品、分离纯化、终产品、制剂、包装和储运都可能成为关键控制点。
第五,更强调药学概念。总颗粒数或总蛋白量不是最终答案,强度、效价、活性成分、功能学验证和批间一致性才是治疗产品需要面对的核心问题。
第六,检测方法要分阶段选择。研发阶段可以采用高通量测序、蛋白质组学、脂质组学和多种正交方法进行深度表征;但放行检测阶段,更需要高效、稳健、可验证、可重复的方法。例如,RNA测序适合机制研究,而针对特定RNA的qPCR可能更适合常规放行检测。
总结下来,EV质量评价追求的不是“最先进的方法”,而是“最适合产品质量决策的方法”。
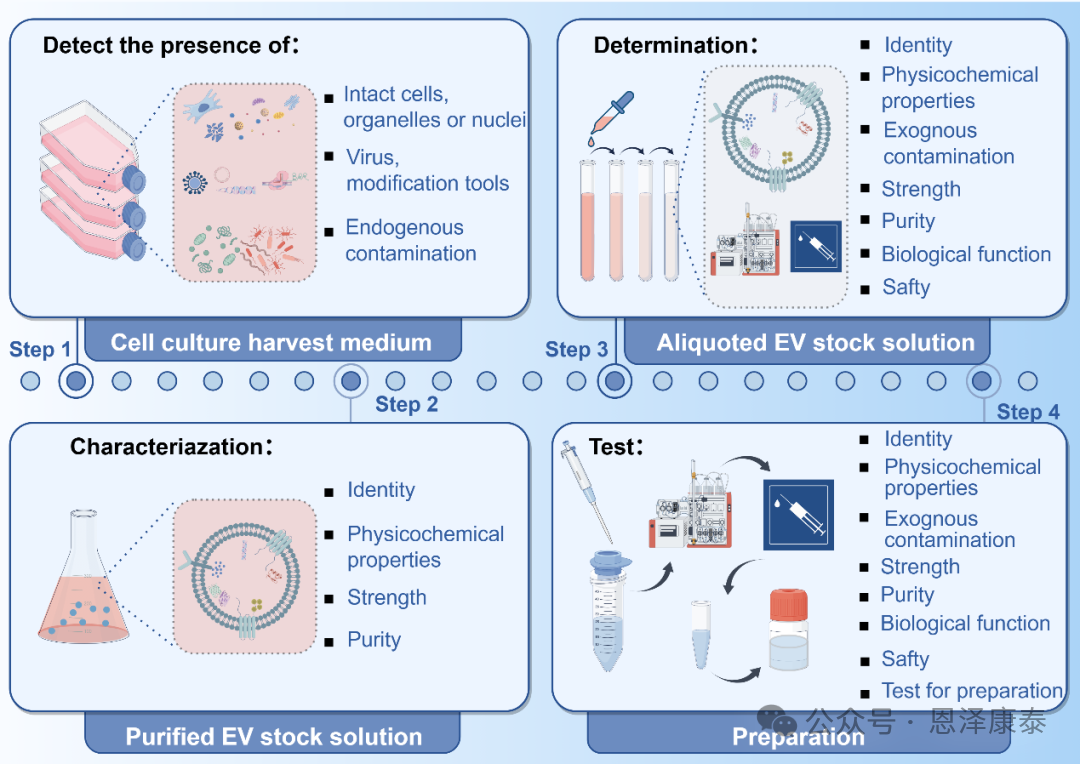
Fig. 4. 文章提出EV生产过程中的质量控制策略,覆盖细胞库、培养上清、分离纯化、中间品、终产品和制剂等关键节点。
关键内容8. 文章提出了候选CQA框架:推动EV质量评价从“经验判断”走向“体系化设计”
文章最后总结了EV治疗产品常见的候选关键质量属性(CQAs),包括:
身份鉴定:阳性标志物、阴性标志物、来源鉴定。
理化性质:粒径分布、Zeta电位。
外源性污染:内毒素、无菌、支原体等。
强度:颗粒浓度、指定粒径范围内颗粒浓度、完整膜颗粒浓度。
纯度与杂质:膜完整性、稳定性、宿主细胞蛋白、残留核酸等。
效价:活性成分、功能实验。
安全性:凝血、免疫原性、促瘤相关风险等。
制剂相关指标:pH、渗透压、辅料残留、粒径、颗粒数、膜完整性等。
需要特别强调的是,文章并没有把这些指标作为“一刀切”的固定标准,而是强调应根据产品类型、细胞来源、生产工艺、作用机制、适应症和风险特征进行个案化设计。
这也是整篇文章最重要的科学态度:
外泌体治疗产品不能照搬一个万能标准,而应建立基于风险、基于机制、基于证据的质量评价体系。
引用文献:
1 Xu, G. et al. Extracellular vesicle-based drug overview: research landscape, quality control and nonclinical evaluation strategies. Signal Transduct Target Ther 10, 255, doi:10.1038/s41392-025-02312-w (2025).
2 Na, T. et al. Quality Evaluation Considerations for Stem Cell-Derived Extracellular Vesicles-Based Therapeutic Products in China. J Extracell Vesicles 15, e70297, doi:10.1002/jev2.70297 (2026).
原文下载:
https://isevjournals.onlinelibrary.wiley.com/doi/10.1002/jev2.70297
(点击文末“阅读原文”可下载原文)
▼
推荐阅读
▼
MISEV2023共识解读合辑
外泌体药物重磅综述发布!中检院首揭中国EV药物监管破局路线
国家队入场!中检院开展外泌体质检,外泌体产业迎来新纪元
干细胞外泌体监管办法:药?械?妆?中检院最新发布终于明确了!
关于恩泽康泰

北京恩泽康泰生物科技有限公司成立于 2017 年,坐落于被誉为“中国药谷”的北京大兴生物医药产业基地,是致力于外泌体技术开发与临床 转化的创新型高科技公司。公司建有外泌体多维分析检测平台Exoomics®、工程化外泌体平台Echosome®及外泌体CMC平台Echopharm®三大技术平台,可提供外泌体科学研究服务、外泌体药物开发、外泌体CRDMO服务及原料生产。公司成立以来累计获得多轮数亿元融资,现已建成超 4000 平米的外泌体 GMP 生产车间,年产能超百万支,并与国内外2000+医院、科研院校及生物企业建立合作,合作发表论文300+篇,处理不同类型的样本10万+例,申报国内外发明专利50+件,参与外泌体国家重大专项及国家重点研发计划专项。获有“国家级专精特新小巨人企业”、“博士后科研工作站”、“高新技术企业”、“中关村金种子企业”、北京市知识产权试点单位”等荣誉。
公司致力于通过提供系统的外泌体研究与应用解决方案,持续的创新和严格的质量要求,让外泌体科技推动医学发展,造福美好生活!